New: Academic Computing Drop-in Sessions
Academic computing drop-in sessions are now run by members of our team on Wednesday afternoons! Head to our drop-in info page for details & schedule....
(thise document is outdated, we are now gradually migrating sections of this document to separate subpages and we are updating them in the new location)
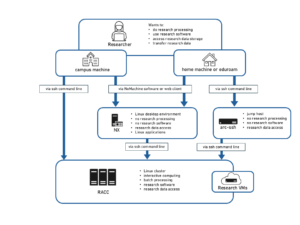
The Reading Academic Computing Cluster (RACC) (formerly the Free Cluster) provides resources for interactive research computing and batch job submissions to the University of Reading staff and students.
The so called “met-cluster” is still running but eventually it will be switched off. More instructions on this cluster are available here. RACC is created to replace all the functions of met-cluster, including interactive computing, batch computing and cronjobs. Met-cluster users should migrate to RACC now. Information relevant for the users migrating from met-cluster are now collected in a separate article: Migrating from met-cluster to RACC “maths-cluster” is going to be merged with RACC as well.
You can log into the Academic Computing Cluster with the following command:
The flag ‘-Y’ might be needed to enable X11 forwarding, to allow running GUI applications. To connect from a Windows computer you will need an ssh client e.g. MobaXterm (note the Portable edition which does not require administrator rights). To connect from Mac you can use Terminal, and ssh will work out of the box, but you will need to install XQuartz for X11 forwarding to work. If you are using Linux, including our Linux Remote Desktop Service, ssh and X11 are already there. See also Access to ACT Servers via an SSH Client.
I you need to connect from an off-campus machine, the available options are listed here.
From the University Wi-Fi network Eduroam access to the UoR services is somewhere between the trusted campus wired network and untrusted off-campus network. In theory, you should be able to ssh directly to RACC when connecting from Eduroam. In practice there might be difficulties, depending on where you are on campus. In such cases you can use one the options for connecting from outside of campus. One of known problems is that internal DNS is not available, in such cases using the IP address 10.210.14.253 instead of the name cluster.act.rdg.ac.uk will help.
cluster.act.rdg.ac.uk (a.k.a. racc-login.act.rdg.ac.uk) acts as a load balancer and it will connect you to the least loaded login node. Both CPU load and memory use are considered. Currently we have 5 login nodes. Those login nodes have decent multicore CPUs and 256GB of memory each and are suitable for low-intensity interactive computing, including running application like Matlab, IDL, etc.
However, you need to remember that those nodes are shared and do not oversubscribe the resources. Bigger jobs should be run in batch mode but. If justified, launching a GUI application in a compute node allocation, such that it cannot disturb other users, is also possible. More information about interactive use of compute nodes is provided in the further sections of this article.
Login node sessions are limited to 4 CPU cores and 100 GB of memory per user. The CPU limit is enforced in such a way that all your processes will share just 4 cores. Regarding the memory limit, if your processess exceed the allocation one of the processes will be killed.
If you notice that the login node becomes slow, you can try disconnecting and connecting to another node, selected by load balancing. Session time and per process time and memory limits might be introduced in the future. Background jobs and cronjobs are not permitted on login nodes.
After the migration in July 2018, the new Unix home directories can now provide more storage space. Also, there are now no technical reasons preventing them to be used for small scale research data processing. More storage space for research data can be purchased from Research Data Storage Service.
The storage volumes are mounted on /storage . This includes /storage/shared/glusterfs and /storage/shared/metcloud, on met-cluster known as /glusterfs and /export/metcloud. Other legacy Met and CINN storage volumes and mount paths will not be supported on the RACC.
300 TB of free scratch space is provided at /scratch1 and /scratch2 . Data in scratch space is not protected and it might be periodically removed to free up space. Scratch storage should only be used for data you have another copy of. User directories will be created on request, when using scratch and not other types of data storage is justified.
Software installed locally is managed by environmental modules, see https://research.reading.ac.uk/act/knowledgebase/accessing-software-on-the-cluster/. These are not the same modules as those available on met-cluster.
Software compiled locally with with the gnu compiler suite will be compiled with the compiler installed with the operating systems, i.e. with gcc 4.8.5. In addition to the packages built with the gnu compiler, some libraries will be also prepared for use with the Intel FORTRAN compiler.
Some scientific software packages are installed from rpms, and do not require loading a module.
A list of installed packages can be found here.
See also: Python on the Academic Computing Cluster and Running Matlab Scripts as Batch Jobs.
Batch jobs is where lies the strength of the cluster. They should be used whenever it is possible, and interactive jobs on the login nodes are only justified for truly interactive work like data visualization or code debugging.
The cluster resources are managed using SLURM workload manger, see https://slurm.schedmd.com/ for more information. Users who are accustomed to the met-cluster and maths-cluster, which use the Grid Engine resource manager will need to familiarize themselves with the new job submission procedures.
SLURM has some commands which will look familiar to Grid Engine users, like qsub, qdel, qstat and qalter. However, they are compatible with PBS (another batch scheduler), not Grid Engine.
Partitions:
In SLURM, partitions are (possibly overlapping) groups of nodes. Partitions are similar to queues in some other batch systems, e.g. in SGE on met-cluster and maths-cluster. The default partition is called ‘cluster’ and it is has 24 hours default time limit, and the maximum time limit is 30 days, but we do not recommend running jobs that long. There is also the ‘limited’ partition, with maximum time limit of 24 hours. The partition ‘limited’ allows access to some of the ‘project’ nodes.
The cluster currently consists of over 50 compute nodes, with core counts varying between 8-cores up to 24-cores per node. There is also a node with 4 GPU devices.
Time and memory limits:
The default time limit in the ‘cluster’ partition is 24 hours and the default memory limit is 1 GB per CPU core. The maximum time limit is 30 days, and the maximum memory limit is not set, it is limited only by the hardware capacity. Users are expected to properly estimate their CPU and memory requirements. Over-allocating resources will prevent other users from accessing unused memory and CPU time. Also, previously consumed resources are used to compute user’s fair-share priority factor, and overprovisioning jobs will have a negative effect on future user’s job priority.
The CPU and memory limits will be strictly enforced by the scheduler. Tasks will be limited to run on the requested number of CPU cores, e.g. if you request one CPU core and run a parallel job, all your threads or processes will run on a single CPU core. Processes, which exceed their memory allocation will be killed by the scheduler.
Fair share policies:
Jobs waiting to be scheduled are ordered according to a multifactor priority. The fair-share component is based on
and the job component depends on
Inactive nodes are automatically shut down. The ‘~’ in the node status shown by the command ‘sinfo’, means that the node is switched off. When you submit a job, such nodes will be automatically switched on and there will be only a short delay before the job starts running, to allow the servers to boot. It takes 6 minutes to boot up all the 17 nodes, but it is much quicker when fewer nodes need to be started.
It should be noted that after a node has just powered up, there might be some problems with MPI jobs, related to the automounter and sssd. A workaround is to add a dummy job slice in the batch script, e.g. ‘cd’, which will fail, but it will force automounting the home directory and the production task will run fine.
Batch jobs are run via a job script, which is submitted with the ‘sbatch’ command. A job script consists of two parts: the resource requests, which are the specifications of required CPU cores, running time, memory etc. which start with ‘#SBATCH’ and one or more job steps, which are the tasks to be run. Note that the lines starting with ‘#SBATCH’ are just comments for the shell, wildcards, shell variables etc. are not expanded in those lines. The script itself is a job step. Other job steps can be created with the srun command.
Relying on the default values of the resource request parameters, a job script can be as simple as:
#!/bin/bash ./myExecutable.exe
and it is submitted with the command:
sbatch script.sh
In most cases, we want to customise the job. Consider the following example script:
#!/bin/bash #these are all the default values anyway #SBATCH --ntasks=1 #SBATCH --cpus-per-task=1 #SBATCH --threads-per-core=1 #again, these are the defaults anyway #SBATCH --partition=cluster #SBATCH --account=local #SBATCH --job-name=example_batch_job #SBATCH --output=myout.txt #SBATCH --time=120:00 #(optional, default is 24 hours) #SBATCH --mem=512 #(optional, default is 1024 MB) ./myExecutable.exe
This is a single process (–ntasks=1, –cpus-per-task=1) batch job. The standard output is redirected into the file ‘myout.txt’. It is recommended that custom run time and memory requirements are specified. For the ‘cluster’ partition the default time is 24h, and the default memory per CPU core is 1024 MB. A job which exceeds those requirements will be killed. Specifying lower requirements which are suitable for your job will decrease the time your jobs is waiting in the queue for resources to become available. In the above example we know that the running time will not exceed 2 hours and set the running time to 120 minutes. As we need less memory than the default 1024 MB, we specify the memory requirements to be 0.5 GB (512 MB). Setting the memory requirements as low as possible increases the chances your job is started quickly and on the fastest CPUs.
Email notifications are disabled by default, they can be enabled with the following directives:
#SBATCH --mail-type=ALL #SBATCH --mail-user=<your email address>
An example script can be found here:
/share/slurm_examples/serial
You might find it confusing that ‘squeue’ will often show that your serial job uses not 1 but 2 CPUs. This is because of hyperthreading. Modern processors have a certain number of physical cores, and might have a larger number of virtual CPUs. Typically one physical core is shared by two virtual CPUs. Slurm counts logical CPUs, not physical cores. However, in most cases of numerical modeling jobs, it doesn’t make much sense for two jobs to share the same physical core; hence here a whole physical core, i.e two virtual CPUs, are allocated to a job.
Jobs arrays allow to easily manage large collections of similar jobs, e.g. to run the same program on a collection of input files, on a range of input parameters, or to run a number of independent stochastic simulations for statistical averaging. We strongly recommend using job arrays whenever possible, because it reduces the load on the scheduler.
Submitting a job array is as simple as adding ‘–array=<index range>’ to the job submission parameters. We can specify the task IDs as a range of consecutive integers: ‘–array=1-10’, a list: ‘–array=1,3,5’, or with an increment ‘–array=1-10:2’.
In the following example we submit a job consisting of 100 array tasks:
#!/bin/bash
#SBATCH --ntasks=1
#SBATCH –-array=1-100
#SBATCH --job-name=example_job_array
#SBATCH --output=arrayJob_%A_%a.out
#SBATCH --time=120:00 #(120 minutes)
#SBATCH --mem=512 #(512 MB)
echo “This array task index is $SLURM_ARRAY_TASK_ID”
./myExecutable.exe input_file_no_${SLURM_ARRAY_TASK_ID}.in
The job tasks are distinguished by their ID provided in the environment variable SLURM_ARRAY_TASK_ID; ‘%A’ and ‘%a’ evaluate to the job ID and task ID, and can be used to construct output file names.
Overview:
Available cluster resources can be displayed with the command sinfo
Running jobs can be displayed with the command squeue (better with -l flag)
Further, job accounting data can be obtained with the command sacct
Batch jobs are submitted using the command sbatch
The commands salloc and srun allow to interactively run tasks on the compute nodes (this is not an interactive session known by met-cluster users)
Jobs can be killed with scancel
Monitoring cluster resources with ‘sinfo’:
As ‘cluster’ is the default partition, it is convenient to display the resources for just this one, by adding ‘-p cluster’ to the ‘sinfo’ command. By default, the nodes which are in the same state are grouped together.
$ sinfo -p cluster PARTITION AVAIL TIMELIMIT NODES STATE NODELIST cluster* up infinite 7 idle~ compute-0-[5-11] cluster* up infinite 1 mix compute-0-0 cluster* up infinite 4 alloc compute-0-[1-4]
The above output shows that nodes 5-11 are idle, and ‘~’ means they are switched off to save power. Nodes 1-4 are fully allocated, meaning they will not be available for new jobs until the jobs currently running on them are finished. The mix state for the compute-0-0 node means that some of the cores on the node are in use and some of them are free.
Further details can be displayed using the ‘-o’ flag. See the manual page, ‘man sinfo’, for more details on format specifiers. In this example, the number of CPU cores are displayed with the command:
$ sinfo -p cluster -o "%P %.6t %C" PARTITION STATE CPUS(A/I/O/T) cluster* idle~ 0/112/0/112 cluster* mix 8/8/0/16 cluster* alloc 64/0/0/64
A/I/O/T stands for Allocated/Idle/Other/Total. The idle and switched off nodes have 112 cores available. There is a node with 8 cores allocated, and another 8 cores idle. In total, there are 120 cores (8 + 112) available for new jobs.
Nodes can be listed individually by adding the ‘-N’ flag:
$ sinfo -p cluster -N -o "%N %.6t %C" NODELIST STATE CPUS(A/I/O/T) compute-0-0 alloc 16/0/0/16 compute-0-1 alloc 16/0/0/16 compute-0-2 alloc 16/0/0/16 compute-0-3 alloc 16/0/0/16 compute-0-4 idle~ 0/16/0/16 compute-0-5 idle~ 0/16/0/16 compute-0-6 idle~ 0/16/0/16 compute-0-7 idle~ 0/16/0/16 compute-0-8 mix 8/8/0/16 compute-0-9 idle~ 0/16/0/16 compute-0-10 idle~ 0/16/0/16 compute-0-11 idle~ 0/16/0/16
Monitoring jobs with squeue
A more informative output formatting can be achieved with the follwing
squeue -o “%.18i %.8u %.8a %.9P %q %.8j %.8T %.12M %.12l %.5C %.10R %p
it might be a good idea to add this as an alias in your .bashrc.
In the SGE resource manager on the met-cluster and the maths-cluster, a job is simply requested with a number of CPU slots allocated to it. On those clusters it is up to the user to spawn required processes, and it is user’s responsibility to not oversubscribe their allocation. SLURM offers more help and flexibility in starting parallel jobs. It will also forcibly limit the resources available to the job, to those specified in the job allocation.
In SLURM, a job can consist of multiple job slices. A job slice is a command or a script. In the simplest case, the execution of the commands in the job script itself is the only job slice; an example is the serial batch job script above. Other job slices within a batch script can be started using the srun command. Jobs slices can run either in parallel or sequentially within the job allocation.
A task can be interpreted as an instance of a job slice. SLURM can start a number of identical tasks in parallel for each job slice, as specified with the flag ‘—ntasks’ for that job slice i.e. for the job script as a whole, or for the srun call.
A task can have more than one CPU cores allocated, such that the user application can spawn more processes and threads on its own. For example, if we run an openMP multi-threaded job, or an application like R or Matlab, which might be starting more processes or threads on its own, we just request one task to start the application, but with the suitable number of CPUs for this single task.
An example of a multi task job is an MPI job. We just specify the number of tasks we want with ‘—ntasks’ and SLURM will start parallel MPI processes for us. Those tasks might use more than one CPU per task if they are multi-threaded.
To have more job slices we use more calls to the srun command in the job script. For example, we can have a job consisting of a data producing slice, possibly with many parallel tasks, running in parallel with a data collector slice, again with many tasks, and many CPUs per task if needed.
This is an example of 16-way MPI job.
#!/bin/bash #SBATCH --ntasks=16 #SBATCH --cpus-per-task=1 #SBATCH --nodes=1-1 #SBATCH --job-name=test_mpi #SBATCH --output=myout.txt #SBATCH --time=120:00 #SBATCH --mem-per-cpu=512 module load MPI/mpich/gcc/3.2.1 srun cd 2>/dev/null #a workaround, needed when node powers up srun myMPIexecutable.exe
The above script requests16 tasks for 16 MPI processes. Typically it is better to have all the processes runing on the same node, it is requested with ‘–nodes=1-1’. In SLURM, the number of tasks represents the number of instances the command is run, typically done by a single srun command. In the above script, we have 16 identical processes, hence this is a slice with 16 tasks and not just one task using 16 CPU cores.
The mpich library version loaded by the module command is built with SLURM support. The srun command will take care of creating and managing MPI processes, it replaces the mpirun or mpiexec commands.
An example of an MPI program and job script can be found here:
/share/slurm_examples/mpi
Similar like in the case of serial jobs, an MPI process (task) gets access to a whole physical core, when possible, and then it is counted as two CPUs in Slurm.
#!/bin/bash #SBATCH --ntasks=1 #SBATCH --cpus-per-task=16 #SBATCH --threads-per-core=1 #SBATCH --job-name=test_smp #SBATCH --output=myout.txt #SBATCH --time=120:00 #SBATCH --mem-per-cpu=512 export OMP_NUM_THREADS=16 ./a.out
In the above script, one task is requested with 16 CPU cores allocated for this task. The executable can use up to 16 CPU cores for its threads or processes. In a similar fashion, parallel Matlab jobs (not tested) can be launched (with Matlab parallel toolbox, but without Matlab parallel server), or any other applications using multiple CPUs and managing them on their own.
Using ‘–cpus-per-task’ is a bit tricky because of hyperthreading. For Slurm the CPU is a logical CPU (hardware thread). In RACC Slurm is configured to always allocate a whole physical core to a task. But, in case of ‘–cpus-per-task’, we are counting Slurm’s CPUs i.e. in case of processors with hyperthreading these are logical CPUs (hardware threads). On most physical nodes we have there is hyperthreading and there are two logical CPUs per physical core. On the VM nodes the CPUs are hardware threads allocated by the hypervisor. If you are happy to count CPU as hardware threads that’s easy and consistent, in both cases. However, often it is better to run just one thread per physical core, and then some customization of the job, depending on the compute node capability, is needed.
The debug partition is created to allow for instantaneous test runs in situations where all cluster resources are allocated, and a regular job would need to wait for its turn. The jobs submitted from debug partition have ability to suspend other running jobs. However, the suspended jobs will stay in the memory on the node, so suspending will not happen if there is not enough memory to accommodate the new job. The maximum runtime for such test jobs is 30 minutes.
The debug partition can be used with batch jobs. But here, we show some examples illustrating the use of sallock and srun commands to run tasks on the nodes interactively. Let us test it with an MPI program which displays the hostname and the “Hello World” message from each process.
module load gcc module load MPI $ srun --partition=debug -n12 ./a.out host compute-1-3.local node 2 : Hello, world host compute-1-3.local node 4 : Hello, world host compute-1-3.local node 6 : Hello, world host compute-1-3.local node 1 : Hello, world host compute-1-3.local node 3 : Hello, world host compute-1-3.local node 5 : Hello, world host compute-1-3.local node 7 : Hello, world host compute-1-4.local node 8 : Hello, world host compute-1-4.local node 9 : Hello, world host compute-1-4.local node 10 : Hello, world host compute-1-4.local node 11 : Hello, world host compute-1-3.local node 0 : Hello, world
The job inherits the environment, so it was sufficient to load the required modules in the shell on the head node.
With salloc we can allocate resources and then run more tasks on the allocated nodes:
$ salloc -n16 --partition=debug salloc: Granted job allocation 301 $ srun ./a.out $ srun ./b.out ... $ scancel 301 salloc: Job allocation 301 has been revoked.
Please remember to release the allocated resources with scancel!
Running job slices in allocation created with ‘salloc’ is quite similar to running them from a job script submitted with ‘sbatch’.
Standard interactive work should be done on the load-balanced login nodes. Interactive use of compute nodes is only allowed in exceptional cases. This is because allocating a whole node for an interactive session, where typically the CPU stays idle most of the time, would too wasteful. We recommend using exclusive node allocation here because strict memory and CPU limits imposed on user jobs are likely to cause problems with GUI applications. Also, it should be noted that dealing with tasks launched from the login node and running on a compute node can be confusing.
The following commands
[cluster-login-0-1 ~]$ module load matlab [cluster-login-0-1 ~]$ srun --exclusive --mem=0 --pty matlab
Will start Matlab with exclusive use of all the CPU cores (—exclusive), and all the memory (—mem=0) of a compute node. By adding a node list you can either request one of the 8-core nodes, with 48 GB of memory: ‘–w compute-1-[0-4]’, or one of the nodes with 16 cores and 96 GB of memory: ‘–w compute-0-[0-11]’. As this is allocated in the default ‘cluster’ partition, the session will expire in 24 hours. This approach should only be taken if such exclusive use of a node is required; all the CPUs should be used in the Matlab code (e.g. start Matlab parallel pool with parpool(16)). Please ensure that your Matlab session really runs on a compute node and not on the login node.
In a similar fashion one can launch IDL and any other GUI application. It is also possible to have a full interactive session by replacing ‘matlab’ with ‘bash’.
[cluster-login-0-1 ~]$ srun --exclusive --mem=0 --pty bash [compute-0-2 ~]$ gnome-terminal [compute-0-2 ~]$ emacs & [compute-0-2 ~]$ exit exit [cluster-login-0-1 ~]$
‘—pty’ stands for pseudo-terminal. Without this option, you will still see the output from the job running on the compute node, but you will not be able to interact with the job. Here we used single task allocations. If you interactively start a multitask job you will have a number of copies of Matlab launched, or a number of instances of bash started (and the job will fail). When ‘—pty’ is used with a multitask job, you will only interact with the pseudo-terminal started in the first task.